
Wir befinden uns in bewegten Zeiten in denen Covid-Maßnahmengesetzte diskussionslos durch das österreichische Parlament gewunken werden und der Staat mit bedingt zugelassenen Covid-Arzneimitteln die Pandemie bekämpft. Teile der Covid-Verordnungen der Bundesregierung werden durch Gerichte wegen Gesetzwidrigkeit wieder aufgehoben. Grund- und Freiheitsrechte der Bürger, vor allem nicht geimpfter Bürger werden eingeschränkt. Verschiedene Berufsgruppen stehen vor „Zwangsimpfungen“ und „Kündigungen“. In den Schulen herrscht Chaos und Spaltung zwischen Lehrern, Schülern und anordnenden Behörden. Daher ist es nun notwendig verschiedene Sachverhalte öffentlich zu machen und die Bürger zu informieren. Denn wären die Covid-19-Maßnahmengesetze durch die Bundesregierung nicht still im Parlament verlängert worden, wären wohl auch die „bedingten Zulassungen“ der aktuell bedingt zugelassenen Covid-Arzneimittel gesetzlich beendet. Es gilt somit den aktuellen Zustand in Österreich zu beleuchten.

Epidemie- und Covid-19-Maßnahmengesetz diskussionslos bis 31.12.2021 verlängert
Mit einem Antrag der Abgeordneten Gabriela Schwarz, Ralph Schallmeiner, Kolleginnen und Kollegen vom 17.6.2021 wird ein Satz in den Gesetzen verändert, die Covid Maßnahmen ermöglichen:
Dem § 50 wird folgender Abs. 24 angefügt: „(24) § 50 Abs. 20 tritt mit Ablauf des 31. Dezember 2021 außer Kraft.“
Bisher wären die Gesetze mit 30.6.2021, also diese Woche außer Kraft getreten. Das Gesetz wurde offenbar mühelos durchgewunken. Wobei – wo blieb die parlamentarische Opposition? Wieso hat niemand die Begründung hinterfragt? Der Zeitrahmen war sehr knapp, das Gesetz trat 2 Tage vor dem Aus in Kraft. Sonst wären am 1. Juli jegliche Maßnahmen und Einschränkungen ohne gesetzliche Grundlage gewesen. Die veränderten Gesetze sind mit den Unterschriften von Van der Bellen und Kurz mit Datum 28.6.2021 in Kraft getreten. Überraschend ist, dass es dazu im Parlament offenbar keine Diskussion gab. Mit dem 105. Bundesgesetz zur Änderung des Suchtmittelgesetzes, des Epidemiegesetzes 1950 und des COVID-19-Maßnahmengesetzes wurde jeweils deren Ende vom 30.6. auf den 31.12.2021 verlängert.

Covid Impfstoffe mit „bedingter Zulassung“ in der EU und in AUT
Jeder Impfstoffentwickler, der einen Impfstoff in der EU in Verkehr bringen will, muss zunächst eine Marktzulassung für den Impfstoff beantragen. Der Antrag wird bei der Europäischen Arzneimittel-Agentur (EMA) eingereicht, die die Sicherheit, Wirksamkeit und Qualität des Impfstoffs bewertet. Gibt die EMA eine positive Empfehlung ab, kann die Kommission mit der Zulassung des Impfstoffs in der EU fortfahren.
Nach positiver Bewertung in Bezug auf Sicherheit, Qualität und Wirksamkeit durch die Europäische Arzneimittel-Agentur (EMA) erteilte die Kommission eine bedingte Zulassung für bislang 4 Impfstoffe folgender Hersteller für Österreich:
- BioNTech/Pfizer (am 21.12.2020)
- Moderna (am 6.1.2021)
- AstraZeneca (am 29.1.2021)
- Janssen Pharmaceutica NV (am 11.3.2021)
Alle 4 Impfstoffe wurden unter „besonderen Bedingungen“ zugelassen. Das bedeutet, dass weitere Nachweise für den Nutzen des Arzneimittels erwartet werden. Die Europäische Arzneimittel-Agentur wird neue Informationen zu diesem Arzneimittel mindestens jährlich bewerten und, falls erforderlich, wird die Zusammenfassung der Merkmale des Arzneimittels aktualisiert werden.
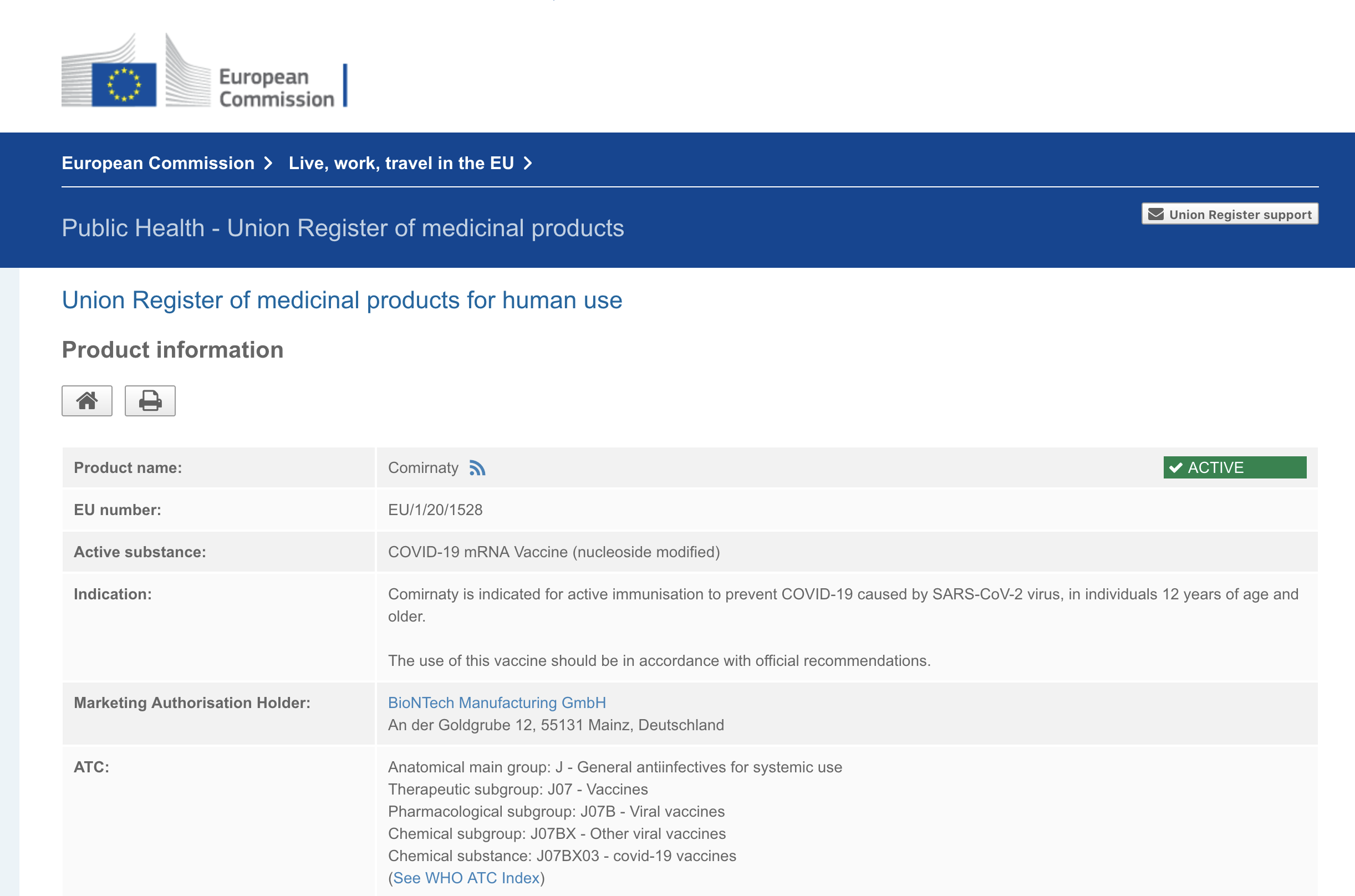
Durchführungsbeschlüsse und Zulassungsbescheide der bedingten Zulassung
finden Sie auf der Website der Europäischen Kommission
Comirnaty
https://ec.europa.eu/health/documents/community-register/html/h1528.htm
Spikevax
https://ec.europa.eu/health/documents/community-register/html/h1507.htm
Vaxzevria
https://ec.europa.eu/health/documents/community-register/html/h1529.htm
COVID-19 Vaccine Janssen
https://ec.europa.eu/health/documents/community-register/html/h1525.htm
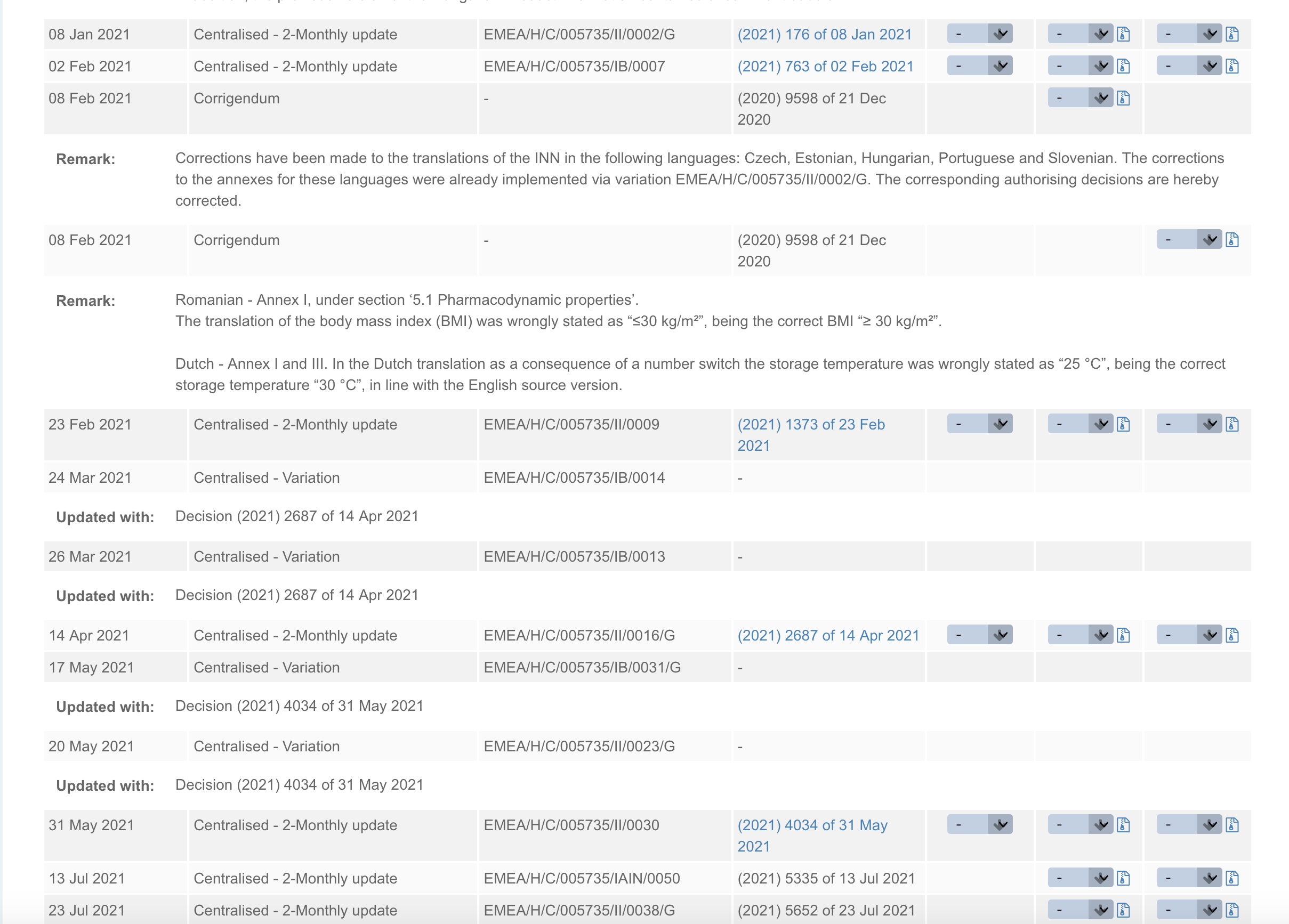
Bedingte Zulassung (Conditional Marketing Authorisation)
Der Antragsteller muss zunächst ausreichend Daten vorlegen, die Qualität, Sicherheit und Wirksamkeit des Impfstoffs belegen und eine Nutzen-Risiko-Bewertung ermöglichen. Bestimmte und zum Zeitpunkt der Zulassung genau zu definierende Daten und Informationen sind nach erfolgter Zulassung zur Begutachtung vorzulegen. Dies erfolgt mit strikten Auflagen und Vorgaben, zu welchen Zeitpunkten die noch ausstehenden Informationen an die Behörden übermittelt werden müssen.
Bedingung für diese Art der Zulassung ist, dass es sich u.a. um eine bedrohliche Erkrankung handelt, für die es derzeit kein optimal geeignetes Medikament gibt. Dies ist bei COVID-19 erfüllt. Beispiel: Am 6. Jänner 2021 hat die Europäische Kommission den Impfstoff der US-amerikanischen Firma Moderna zugelassen, nachdem die EMA eine bedingte Zulassung für Personen ab 18 Jahren empfohlen hatte.
Die Europäische Kommission hat im März 2021 dem von Janssen Pharmaceutica NV, einem Unternehmen der Pharmasparte des Konzerns Johnson & Johnson, entwickelten COVID-19-Impfstoff eine bedingte Zulassung erteilt. Er ist damit der vierte in der EU zugelassene Impfstoff gegen COVID-19.
Eine bedingte Zulassung ist die Zulassung eines Arzneimittels, für das noch nicht alle für eine normale Zulassung erforderlichen Daten vorliegen. Eine solche bedingte Zulassung kann dann erwogen werden, wenn der Nutzen der sofortigen Verfügbarkeit des Arzneimittels die Risiken im Zusammenhang mit der unvollständigen Datenlage deutlich überwiegt. Wie bei allen anderen Impfstoffen und Arzneimitteln stellt sie jedoch auch sicher, dass dieser COVID-19-Impfstoff den EU-Standards entspricht.
Die Daten, auf deren Grundlage die Europäische Arzneimittelbehörde EMA eine bedingte Zulassung bzw. eine Conditional Marketing Authorisation erteilt, müssen jedenfalls zeigen, dass die Vorteile und der Nutzen der sofortigen Verfügbarkeit eines Impfstoffes (oder eines Medikaments) die Risiken für die Patientinnen und Patienten überwiegen.
Wie werden Corona Impfstoffe nach der bedingten Zulassung geprüft?
Sobald neue Arzneimittel auf den Markt kommen, sammeln und überprüfen die Hersteller und EU-Behörden kontinuierlich neue Informationen über Arzneimittel und ergreifen bei Bedarf Maßnahmen. Im Einklang mit dem EU-Sicherheitsüberwachungsplan für COVID-19-Impfstoffe wird diese Überwachung häufiger stattfinden und Aktivitäten umfassen, die speziell für COVID-19-Impfstoffe gelten. Die Unternehmen werden zum Beispiel zusätzlich zu den gesetzlich vorgeschriebenen regelmäßigen Aktualisierungen monatliche Sicherheitsberichte vorlegen und Studien zur Überwachung der Sicherheit und Wirksamkeit von COVID-19-Impfstoffen nach ihrer Zulassung durchführen.
Als Reaktion auf Bedrohungen der öffentlichen Gesundheit wie die derzeitige Pandemie verfügt die EU über ein spezifisches Regulierungsinstrument, das eine frühzeitige Verfügbarkeit von Arzneimitteln für den Einsatz in Notsituationen ermöglicht. In solchen Notsituationen ist das Verfahren für die bedingte Marktzulassung so konzipiert, dass Marktzulassungen so schnell wie möglich erteilt werden, sobald ausreichende Daten vorliegen. Es bietet der EU einen soliden Rahmen für die beschleunigte Zulassung und die Unbedenklichkeit sowie Sicherheitsvorkehrungen und Kontrollen nach der Zulassung.
Für ihre Bewertung wird die EMA alle vom Impfstoffentwickler vorgelegten Nachweise einer unabhängigen, gründlichen und soliden Prüfung unterziehen. Das Verfahren umfasst verschiedene Kontrollen und Gegenkontrollen und beruht auf einem Peer-Review-System, an dem ein Expertenteam beteiligt ist: zwei für die Bewertung zuständige Berichterstatter, ein Gutachter, Fachausschüsse und Arbeitsgruppen (z. B. der Ausschuss für Risikobewertung im Bereich der Pharmakovigilanz (PRAC) für Sicherheit und die Arbeitsgruppe „Biotechnologie” für Qualität) und schließlich der Ausschuss für Humanarzneimittel der EMA (mit Mitgliedern aus allen Mitgliedstaaten), der die Empfehlung ausspricht. Der Ausschuss für Humanarzneimittel wird nur dann eine positive Empfehlung abgeben, wenn die Nachweise überzeugend belegen, dass der Nutzen der Impfung die Risiken überwiegt.
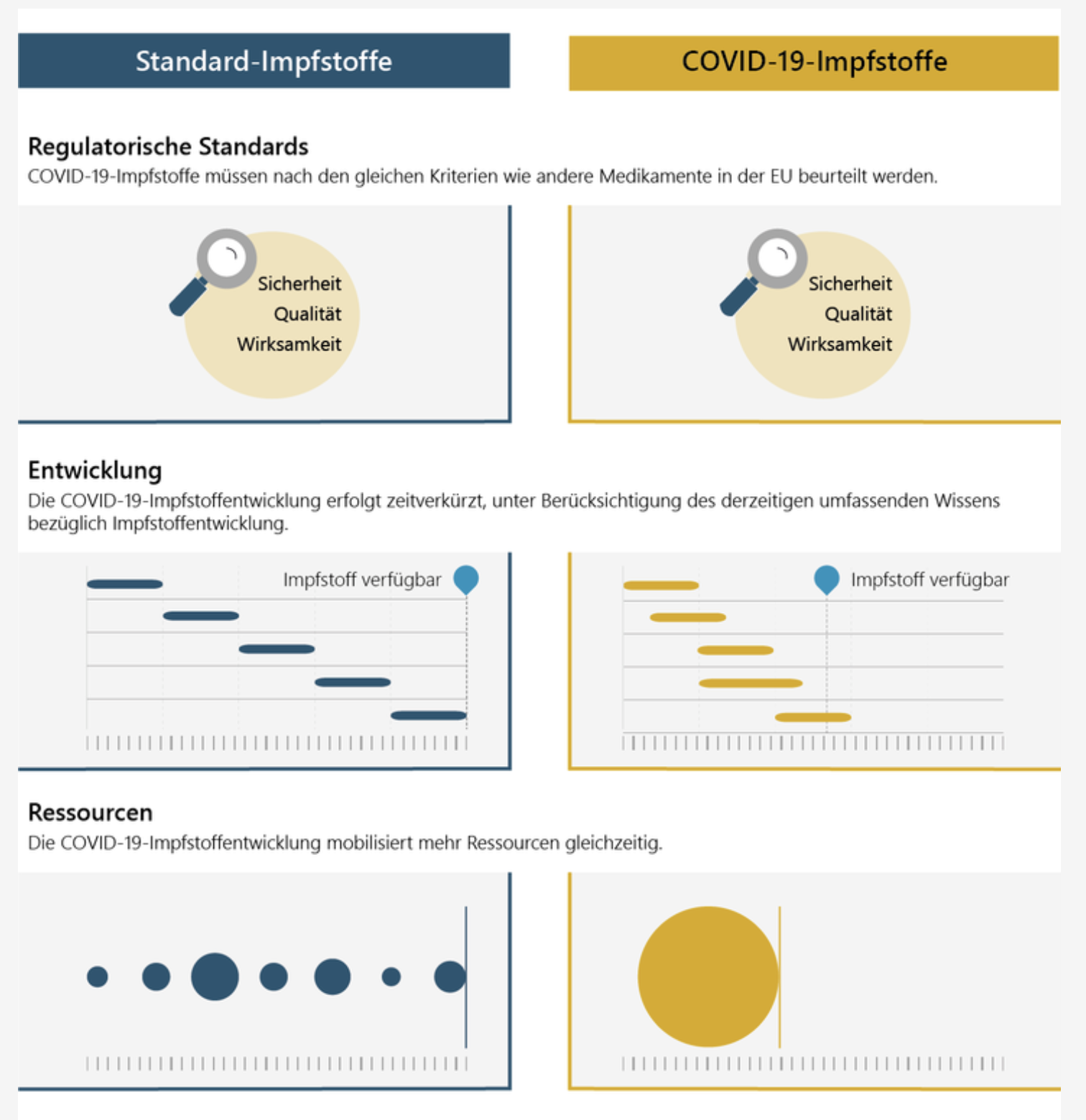
Marktzulassung durch die Europäische Kommission
Die Europäische Kommission ist für die Marktzulassung rechtlich verantwortlich. Nach einer positiven Empfehlung des Ausschusses für Humanarzneimittel (CHMP) der EMA wird die Kommission die Zuverlässigkeit aller Elemente überprüfen, die die Marktzulassung stützen. Dazu gehören wissenschaftliche Begründungen, Produktinformationen, Schulungsmaterial für Angehörige der Gesundheitsberufe, Kennzeichnung, Pflichten des Impfstoffentwicklers, Anwendungsbedingungen und mögliche Verpflichtungen für die Mitgliedstaaten. Die Kommission ist auch dafür verantwortlich, dass Patienten und Angehörige der Gesundheitsberufe in der gesamten EU über alle erforderlichen Informationen in ihrer Landessprache verfügen.
Bevor sie ihren Beschluss fasst, konsultiert die Kommission die Mitgliedstaaten, die für die Vermarktung und Verwendung des Produkts in ihren Ländern zuständig sind (per Komitologieverfahren – Prüfverfahren). Wenn die Mitgliedstaaten die Zulassung mit qualifizierter Mehrheit befürworten, kann die Kommission mit der Annahme ihres Beschlusses zur Genehmigung der Vermarktung des Impfstoffs fortfahren.
Danach darf der Impfstoff überall in der EU in Verkehr gebracht werden. Impfstoffentwickler brauchen keine weiteren Zulassungen in den verschiedenen EU-Mitgliedstaaten zu beantragen.
Die Kommission genehmigte den Vertrag mit Janssen am 8. Oktober 2020
Auf der Grundlage der befürwortenden Stellungnahme der EMA hat die Kommission vor Erteilung der bedingten Zulassung alle dieser Zulassung zugrunde liegenden Elemente überprüft und die Mitgliedstaaten konsultiert.
Nach Erteilung der bedingten Zulassung kann Janssen ab dem zweiten Quartal 2021 beginnen, 200 Millionen Dosen seines in einer Einzeldosis zu verabreichenden Impfstoffs an die EU zu liefern. Gemäß dem Vertrag haben die Mitgliedstaaten die Möglichkeit, weitere 200 Millionen Dosen zu erwerben. Sie kommen zu den 600 Millionen Dosen des Impfstoffs von BioNTech-Pfizer, den 460 Millionen Dosen des Impfstoffs von Moderna und den 400 Millionen Dosen des Impfstoffs von AstraZeneca hinzu.
Fach- und Gebrauchsinformationen der zugelassenen COVID-19-Impfstoffe Neu
Comirnaty (mRNA-Impfstoff) Neu
Zulassungsinhaber BioNTech
Zulassung am 21. Dezember 2020.
Am 08. Jänner 2021 wurde eine geänderte Fachinformation für Comirnaty zugelassen, nach welcher sechs (anstatt ursprünglich fünf) Dosen pro Behälter entnommen werden können.
Am 30. März 2021 wurde die kurzfristige Lagerung bei -25 °C bis -15 °C zugelassen (siehe Fachinformation Punkt 6.3).
Am 28.Mai 2021 erweiterte die EMA das Anwendungsgebiet „zur aktiven Immunisierung von Personen ab 12 Jahren“.
Die Produktinformation wurde mit „Stressbedingte Reaktionen“ in 4.4 und „ausgedehnte Schwellung der geimpften Gliedmaße“ und „Schwellungen im Gesicht“ zu 4.8. aktualisiert.
Gebrauchsinformation (29.07.2021)
Fachinformation (29.07.2021) für medizinisches Fachpersonal
Spikevax (mRNA-Impfstoff) – vormals Moderna Neu
Zulassungsinhaber Moderna
Zulassung am 6. Jänner 2021
Änderung vom 22.06.2021: Die Bezeichnung des Impfstoffes Moderna wurde mit 22.06.2021 auf SPIKEVAX geändert.
Indikationserweiterung auf ab 12 Jahre.
Gebrauchsinformation (26.07.2021)
Fachinformation (26.07.2021) für medizinisches Fachpersonal
Vaxzevria (Adenovirus-Impfstoff) – vormals AstraZeneca Neu
Zulassungsinhaber AstraZeneca
Zulassung am 29.Jänner 2021
Am 29.03.2021 wurde der Name von AstraZeneca auf Vaxzevria Injektionssuspension geändert.
Die Änderung betrifft die Möglichkeit von Guillain -Barré Syndrome (GBS) nach Impfung (4.4).
Gebrauchsinformation (19.07.2021)
Fachinformation (19.07.2021) für medizinisches Fachpersonal
Janssen (Adenovirus-Impfstoff) Neu
Zulassungsinhaber Janssen-Cilag
Zulassung am 11. März 2021
Die Änderung betrifft die Möglichkeit von Guillain -Barré Syndrome (GBS) nach Impfung (4.4 und 4.8).
Gebrauchsinformation (22.07.2021)
Fachinformation (22.07.2021) für medizinisches Fachpersonal
Quellen:
https://www.basg.gv.at/konsumentinnen/wissenswertes-ueber-arzneimittel/covid-19-impfstoffe
https://www.ages.at/themen/krankheitserreger/coronavirus/entwicklung-und-zulassung-von-impfstoffen/
https://ec.europa.eu/commission/presscorner/detail/de/ip_21_1085
Informationen zur Bedingten Zulassung (Conditional Marketing Autohrisation) und zu beschleunigten Zulassungsverfahren finden Sie auf folgender Website der AGES sowie auf der Website der EMA:
https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/accelerated-assessment
Epidemie- und Covid-19-Maßnahmengesetz diskussionslos bis 31.12.2021 verlängert
Für alle Firmen, Institutionen, Personen und überhaupt „Jeden und „Alles“ gilt die Unschuldsvermutung. (hu) ++ende++
 |
Herbert Unger – freier Journalist bei bkftv.at herbert.unger@bkftv.at oder 06645344908 Meine Artikel: https://bkftv.at/author/hu/ Schreiben Sie mir zum Thema Vertrauliche Kommunikation über: Threema ID hcclnoname: WUU3ZJJV Signal-Messenger oder persönliche Treffen: Face-to-Face |





